Preparing the P450 system
Note
The scripts for this step of the tutorial can be found in
p450_enzyme/1_solvation.
Introduction
The cytochrome P450 family of haem-containing enzymes is found in all kingdoms of life. In humans, P450 enzymes are important for the synthesis of hormones and drug metabolism among other functions. In this tutorial we will explore how to model P450cam (also known as CYP101), a bacterial P450 which catalyses the hydroxylation of camphor. P450cam was the first P450 to have its structure solved by X-ray crystallography and has been the subject of extensive experimental and modelling studies.
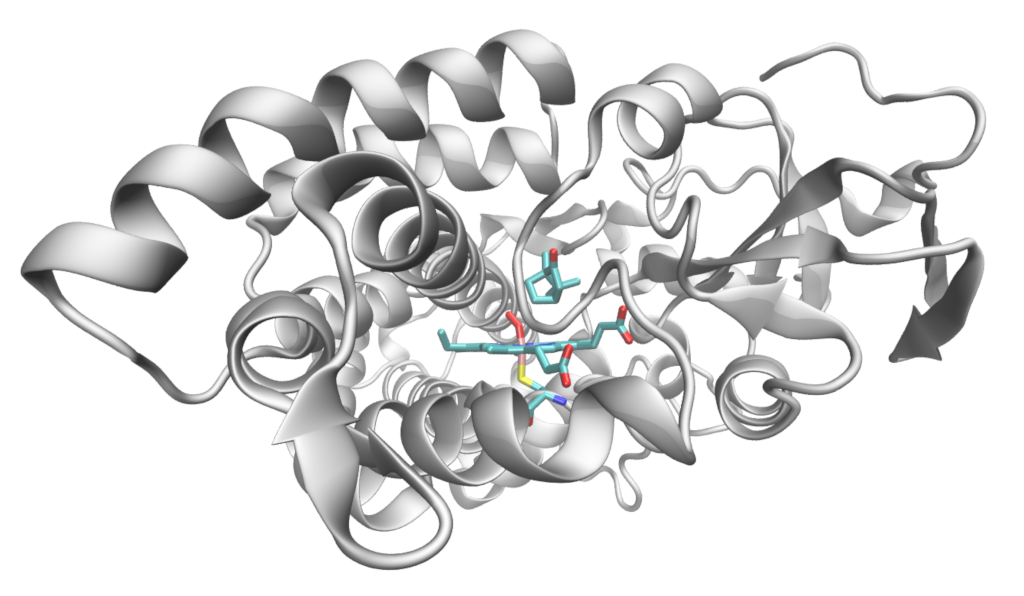
Structure of P450cam showing the haem-oxygen complex, bound cysteine residue and camphor substrate.
QM/MM methods are particularly well-suited to enzyme modelling because they divide naturally into an active site, which should be modelled at the quantum mechanical level to give an accurate description of reactivity, and the protein environment which can be described using standard biomolecular forcefields. In this tutorial, we will use DFT for the quantum mechanical region and the CHARMM forcefield to describe the MM enviroment.
In the first part of the tutorial, we demonstrate how to set up the P450cam system so that is ready for QM/MM modelling, starting from an X-ray structure, introducing a solvent environment and performing classical molecular dynamics to equilibrate the system.
Initial preparation of the system
We start from a P450cam structure from the Protein Data Bank (PDB) with the PDB code 1DZ8. This is an oxygen complex of P450cam including the substrate camphor.
Initial preparation of the system is necessary to transform a raw X-ray
crystallography structure into a form suitable for computational modelling.
The first stage of the process is in the ChemShell script initial_structure.py.
In this tutorial we have provided the P450 structure as a PDB file 1dz8.pdb.
Before you run the ChemShell script, open the PDB file in a visualiser such
as VMD and familiarise yourself
with the X-ray structure.
The PDB file is read into ChemShell in the usual way:
# Read in the P450cam X-ray structure from disk
p450_original = Fragment(coords='1dz8.pdb')
Note
Alternatively, ChemShell can download the structure from the PDB using the following command:
p450_original = Protein(id='1DZ8', source='rcsb')
Note that ChemShell will process the raw PDB file when it is read in to make it more suitable for computational studies:
PDB residue names will be renamed to conform to CHARMM conventions (in this case, potassium ions will be renamed to POT, and the oxygen molecule to O2)
The components of the system will be sorted into PROT (the protein, including haem complex), MISC (camphor and other molecules) and CW (crystal water) segments, useful for later processing.
Where two alternative positions for an atom are given in the PDB file, ChemShell will select the most common based on consideration of mean occupancy and b-factors. A summary table of the choices made will be printed.
Warning
The example PDB crystal structure is sufficiently complete to be used for modelling, but in general PDB structures should be carefully checked for missing structural information. If there are missing residues or loops, user intervention is required to make sure the protein chain is complete before it is loaded into ChemShell.
Once the PDB structure has been loaded into a Fragment object, we can use ChemShell commands to manipulate it further. We first need to isolate one of the chains of the protein, to obtain the biological assembly (i.e. the functional form of the molecule). In our example we choose chain A, as it contains the bound oxygen molecule already. The chain can be selected using an atom selection command:
# Create a new Fragment containing only chain A, which includes bound O2
indices_chainA = p450_original.select(chainIDs='A')
p450 = p450_original.getSelected(indices_chainA)
Note
If molecular oxygen were not already present (as in chain B, for
example), we can also ChemShell to bind an oxygen molecule to the haem
using the bind method. An example script p450_bind_o2.py is
provided to illustrate this.
We next remove unwanted species which have been artifically introduced during the crystallisation process, including a Tris buffer molecule and potassium ions:
# Delete Tris buffer
p450.delete(residues=['TRS'])
# Delete (potassium) ions which are artefacts of crystalisation
p450.deleteIons()
A list of atoms and coordinates that have been deleted will be printed when these commands are run.
Next we check that the segments of the new fragments are as expected,
using the unique function from the NumPy mathematical library, and
assign a title to the new Fragment:
# Check the segments of the new fragment
print("\n *** p450 segments =\n", unique(p450.segments))
# Give the new system a title
p450.title = 'p450'
Finally, we save the new Fragment as a PDB file, ready for the solvation stage:
# Save the resulting PDB
p450.save('p450_a.pdb')
After running the script, open the resulting p450_a.pdb file in your
visualiser and compare it to the original 1dz8.pdb.
The P450cam system is now ready for the solvation workflow. Note that further preparation is required (for example, hydrogen atoms are still missing), but this will be handled by the workflow itself in the next stage.