Hybrid delocalised coordinates
Introduction
Generally, the representation of a chemical system in internal coordinates is
more efficient for geometry optimisation than its representation in Cartesian
coordinates.
Among the various kinds of internal coordinate representations (e.g. Z-matrix,
natural internal coordinates, delocalised internal coordinates), the
delocalised internal coordinates are most decoupled from each other
since they are a non-redundant set of linear combinations of a set of coupled,
redundant primitive internal coordinates such that the spectroscopic G matrix
becomes diagonal.
Since the CPU requirements of the diagonalisation of the G matrix scales as the
number of atoms cubed, the system can be split up into
residues within which the internal coordinates are
defined and delocalised.
If more than one residue is defined, or if some atoms are represented using
Cartesian coordinates, Cartesian coordinates of all atoms of each residue are
added to the redundant set of coordinates to be delocalised such that the
delocalised coordinates include information about the relative position and
orientation of the residues.
We will call these coordinates hybrid delocalised internal coordinates
(HDLC).
The redundant set of primitive internal coordinates is generated in two ways:
 |
 |
| Primitive internal coordinates
(default, recommended for geometry optimisation and the environment of a
reaction in a transition state search):
the angles, linear angles, improper dihedrals and dihedrals are defined based
on connectivity from covalent radii and shortest distance branched path |
Total connection scheme
(recommended for the reaction core in a transition state search):
every atom is connected to every atom, no angles and dihedrals are defined |
Overview of an optimisation using hybrid delocalised coordinates:
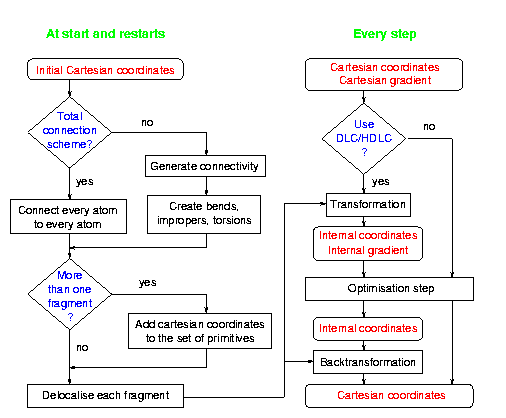
HDLCs in DL-FIND
The HDLC optimiser uses Cartesian coordinates (Fragment tag
) for all
input / output.
It converts the part of the system to be represented in HDLC automatically
"on the fly".
All atoms being part of a residue defined using the
argument residues= are optimised using HDLC, the other atoms are
optimised using Cartesian coordinates.
The coordinates are delocalised only within each residue.
Definition of hybrid delocalised internal coordinates
By default, DL-FIND uses Cartesian coordinates for geometry optimisation
and transition state search for all atoms.
- The atoms whose position is to be represented using
HDLCs must be explicitly assigned to a
residue.
- The coordinates are delocalised over each residue only.
- The atoms not being part of any residue are represented using Cartesians.
- If one single residue spans all atoms, the 6 external degrees of freedom
are retained. For pure internal delocalised coordinates, the dlc
option should be used.
The primitive internal coordinates to be delocalised are generated
automatically. Internal coordinates to be constrained are added automatically
if required.
Definition of residues
Format of the residue membership list
{{residue names} {{atoms of residue1} {atoms of residue2} ...}}
The atoms are counted from 1. Example:
{{res1 res2 res3} {{1 2 3 4 5} {6 7 8} {9 10 11}}}
The inlist command
The inlist command manipulates residue membership lists based on set operations
and returns the resulting residue membership list.
It provides five functions:
| Function |
Arguments |
Opt. arguments |
Description |
| combine |
residues, target, sets |
mode, (set1, set2 instead of sets) |
Combine two or more residues to one residue |
| intersect |
residues, target, sets |
mode, (set1, set2 instead of sets) |
Intersect two or more residues to one residue |
| exclude |
residues, target, set1, set2 |
- |
Exclude atoms of set2 from set1 to target, keep set2 |
| merge |
residues, residues2 |
- |
Merge residue definitions from two
lists. Residues2 preceeds. |
| remove |
residues, sets |
- |
Remove residue sets from a residue list |
| pick |
residues, sets |
- |
Return a list containing only selected sets |
The command inlist accepts these arguments:
| Argument |
Argument type |
Functions |
To specify |
| function= |
string |
- |
Allowed operations: combine, intersect, exclude, merge, remove |
| residues= |
residue list |
all |
Residue membership list to start from |
| sets= |
list |
combine, intersect, remove |
Names of residues to perform set operation on
(see note 1) |
| set1= |
string |
combine, intersect, exclude |
Name of the first residue to perform set
operation on (see note 2) |
| set2= |
string |
combine, intersect, exclude |
Name of the second residue to perform set
operation on (see note 2) |
| target= |
string |
combine, intersect, exclude |
Name of the resulting residue |
| residues2= |
residue list |
merge |
Second residue membership list |
| mode= |
string |
combine, intersect |
Keep ("add") or remove ("remove") the original
sets (default: remove) |
Note 1: For the functions combine and
intersect, either set1= and set2= or sets=
can be specified
Note 2: For the function exclude, atoms of
set2 are excluded from set1, set2 is kept and the
resulting set is stored as target
Other commands for residue membership list manipulation
| Command |
Arguments |
Opt. arguments |
Description |
| res_selectall |
coords= |
- |
Return a residue list defining one residue
containing all atoms |
| pdb_to_res |
name of the PDB file |
names=, excluded_names=
(note 1) |
Return a residue list according to a PDB file
(see note 2) |
| select_residue |
residues=, residue=, coords=, result= |
- |
Copy the atoms of the specified residue of a residue from the fragment coords= to the fragment result= |
Note 1: If names=(list) is present, only
residues are included whose residue name field matches one of the names given
in the list, if exclude_names=(list) is present, only residues are
included whose name field does not match any of the names given in the list.
The argument names= precedes
Note 2: The residue names are composed from the chain and
residue name specification fields if the chain specification is not empty or
from the residue name and residue number specification fields otherwise
Examples of residue definitions
- set residues [ pdb_to_res "1PBE.pdb" names= {FAD PHB} ]
- set residues [ inlist function=combine residues= $from_pdb sets=
{FAD PHB} target=fixed ]
- dl-find coords=c result=res.c coordinates=hdlc residues= [ res_selectall coords=c ]
theory=mndo : hamiltonian=am1
- select_residue coords=1PBE.c result=FAD.c residues= $residues
residue=FAD
HDLC constraints
Cartesian constraints (frozen atoms) are specified using the
residue membership list or via the keyword active_atoms.
If defined using the residue membership list, all frozen atoms must belong to a
residue named "fixed".
Internal constraints (distances, angles and dihedrals) are
specified using the constraints=... argument of the dl-find
command.
For the format, see the examples below.
The atom sequence numbers are counted in the same order as in the input
geometry provided with the coords=... argument and the
residue membership list provided with the
residues=... argument.
No constraint may be defined across a residue boundary.
Use the command inlist function=combine to satisfy this condition.
Examples
- dl-find coords=c result=res.c coordinates=hdlc residues= {{fixed HDLC1} {{1 2 3 6}
{4 5 7 8}}} theory=mndo : hamiltonian=am1
- dl-find coords=c result=res.c coordinates=hdlc residues= $residues constraints=
{{bond 3 5} {angle 3 5 6} {torsion 1 2 3 4} {cart 3 0 0 -1}} theory=mndo
Concerning the cart entry: {cart 3 0 0 -1} fixes the x
component of the 3rd atom, {cart 17 0 0 -2} fixes the y component of
the 17th atom and so on. These are handled as internal constraints, and thus
are only available for HDLC for atoms that are part of a residue.
Practical hints
- Use the argument residues=... and the
commands for the manipulation of residue membership
lists
- Recommended size of a residue is 3 to 50 atoms
- The atoms of the residue named "fixed" are kept fixed
- The argument active_atoms=... can be used if most of the atoms are
to be kept fixed
- Try to cut single bonds only
- Try to avoid cutting rings and other crosslinked subsystems
- Do not put more than one molecule into a residue
- Use the PDB fragmentation for biomolecules
|
